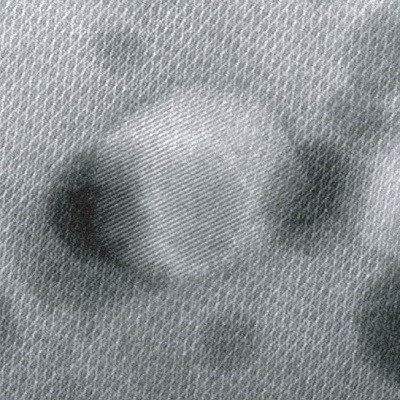
Two years ago, Penn theoretical chemist Andrew M. Rappe visited the lab of Tae-Woo Lee at Seoul National University, and the discussion soon turned to whether they could develop a theory to help explain some of their experimental results. The material they were studying was formamidinium lead bromide, a type of metal-halide perovskite nanocrystal (PNC). Results collected by the Lee group seemed to indicate that green LEDs made with this material were working more efficiently than expected. “As soon as I saw their data, I was amazed by the correlation between the structural, optical, and light-efficiency results. Something special had to be going on,” says Rappe.
PNCs like formamidinium lead bromide are used in photovoltaic devices, where they can store energy as electricity or convert electric current into light in light-emitting devices (LEDs). In LEDs, electrons are carried from an electron-rich (n-type) region to a high-energy level in an electron-poor (p-type) region, where they find an empty lower-energy state, or “hole,” to drop down into and emit light. A material’s efficiency is determined by how well it can convert light into electricity (or vice versa), which depends on how easily an excited electron can find a hole and how much of that energy is lost to heat.

A collaborative study by a team of materials scientists and theoretical chemists demonstrates how a class of electroluminescent materials, key components of devices such as LED lights and solar cells, can be designed to work more efficiently.
To make sense of the Lee group’s results, Penn postdoc Arvin Kakekhani began working with Young-Hoon Kim and Sungjin Kim of Seoul National University to develop a computational model of the material’s unexpected efficiency and to design targeted follow-up experiments to confirm these new theories. “We spent a lot of time cross linking experiment and theory to rationalize every single experimental observation that we have,” says Kakekhani about the research process.
After months of exchanging ideas and narrowing down potential theories, the researchers developed a theoretical model using a method known as density functional theory, a modeling approach that relies on mathematical theories from quantum mechanics. While DFT has been used in the field for many years, the implementations of this theory can now efficiently incorporate the impacts of small, delocalized quantum mechanical interactions, known as van der Waals forces, which are known to play a major role in the behavior of soft materials that are similar to the PNCs used in this study.
Using their new model, the researchers found that the PNCs were more efficient if the size of the quantum dots were smaller, since the probability of an electron finding a hole was much greater. But because reducing a particle’s size also means increasing its surface-to-volume ratio, this also means that there are more places along the material’s surface that are prone to defects, where energy from electrons can easily be lost.
To address both challenges, the researchers found that a simple chemical substitution, replacing formamidinium with a larger organic cation called guanidinium, made the particles smaller while also preserving the structural integrity of the material by allowing more hydrogen bonds to form. Building on this alloying approach, the researchers found additional strategies to improve efficiency, including the addition of long-chain acids and amines to stabilize surface ions and the addition of defect-healing groups to “heal” any vacancies that might form.
As a theoretical chemist, one thing that stood out to Kakekhani was how well the model’s predictions and experimental data aligned, which he attributes in part to using a theory that incorporates van der Waals forces. “You don’t fit parameters that make the theory specific to the experiment,” he says. “It’s more like first principles, and the only knowledge that we have is what type of atoms the materials have. The fact that we predicted the results based on almost pure mathematical operations and quantum mechanical theories in our computers, in close correspondence to what our experimental colleagues found in their labs, was exciting.”
While the current study provides specific strategies for materials that have the potential for widespread use as solar cells and LEDs, this strategy is also something that could be adopted more generally in the field of material science. "Advancement of the Internet of Things and the drive toward optoelectronic computing both demand efficient light sources, and these novel perovskite-based LEDs can lead the way,” Rappe says.
For Kakekhani, this work also highlights the importance of detailed, theory-driven insights for gaining a thorough understanding of a complex material. “If you don’t fundamentally know what is going on and what is the underlying reason, then it is not really extendable to other materials,” says Kakekhani. “In this study, having that long period of trying to rule out theories that didn’t actually work was useful. At the end, we found a really deep reason that was self-consistent. It took a lot of time, but I think it was worth it.”
This work was the result of a collaboration between University-of-Pennsylvania (Penn), Seoul National University (SNU), the Korea Advanced Institute of Science and Technology (KAIST), the Ecole Polytechnique Fédérale de Lausanne (EPFL), the University of Tennessee (UTK), the University of Cambridge, the Universitat de Valencia (UV), the Harbin Institute of Technology (HIT), and the University of Oxford.
Read the original article on University of Pennsylvania.