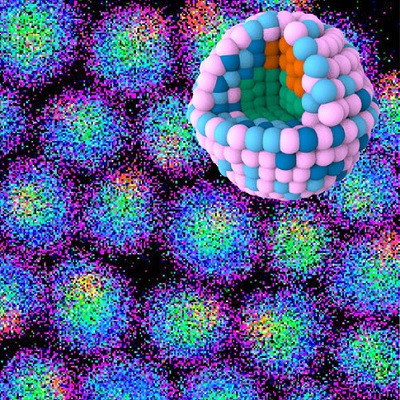
With their tiny, nanoscale-sized pores, the materials are useful for many sustainability applications, but because they are built by chemists in labs molecule by molecule, they are cumbersome and expensive to develop.
A Washington State and Oregon State University research team has developed a unique computer algorithm that plays a game of 20 questions, quickly narrowing down thousands of possible molecular designs to find the optimal one with minimal cost and effort.
“A key challenge is that the nanoporous materials are a mixture of different chemical elements that you have to compose and figure out the best combination,” said Aryan Deshwal, the first author on the study published in the journal, Molecular Systems Design and Engineering Journal.
The nanoporous materials have a huge variety of potential molecular building blocks and arrangements that can be nearly endlessly mixed, said Deshwal, a doctoral student in the School of Electrical Engineering and Computer Science.
“If we were to try out new configurations of these elements and their structures in a laboratory every time, it would be very expensive, so the computational challenge is how to figure out the right combination of elements that have the properties that you care about,” he said. “That’s where our AI-based algorithmic work comes in.”
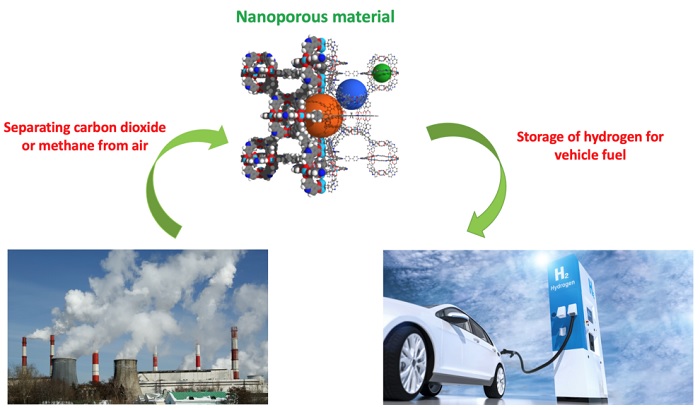
The algorithm the researchers developed narrows down molecular design possibilities for nanoporous materials that can solve challenges in hydrogen fuel storage and carbon or methane capture technology.
As part of the proof-of-concept study, the researchers narrowed down the best candidate for a nanoporous material to absorb methane, a potent greenhouse gas that contributes to global warming. After evaluating just 120 possible candidates, they found the already known best candidate from a library of 70,000 materials which is considerably better than traditional algorithms have performed.
“Aryan’s algorithms are able to find the best material with fewer number of evaluations,” said Jana Doppa, corresponding author on the study and George and Joan Berry Associate Professor in the School of Electrical Engineering and Computer Science. Cory Simon, a leading expert in nanoporous materials research at Oregon State University, was also a co-author.
One of the reasons the algorithm did well is that it looks at the material’s three-dimensional structures themselves.
“We are trying to do a somewhat smarter search, and the existing methods that are used were not trying to exploit models of relationship between structure of material and its properties,” said Deshwal. “We explicitly build statistical models, which allowed us to predict the properties for unknown materials and have well-calibrated uncertainty, which means you know what you don’t know, so when we explored the space, we explored it in a much smarter way rather than randomly.”
As their algorithm came upon each new iteration of the material, it conducted an experiment virtually, updated its understanding about the structure and property relationship, and then, based on that, selected another nanoporous material.
The researchers now aim to further automate and generalize the methodology. They have already made a fundamental advancement towards this goal in a new paper which will be presented at the 2021 Conference on Neural Information Processing Systems (NeurIPS). They hope to use the unique algorithms to improve searches in other types of real-world applications, such as in the design of catalysts that are used in industrial processes. The work was funded by the National Science Foundation.
Read the original article on Washington State University.