Our understanding of biological proteins does not always correlate with how common or important they are. Half of all proteins, molecules that play an integral role in cell processes, are intrinsically disordered, which means many of the standard techniques for probing biomolecules don’t work on them.
Now researchers at Kanazawa University in Japan have shown that their home-grown high-speed atomic force microscopy technology can provide information not just on the structures of these proteins but also their dynamics. The study is reported in Nature Nanotechnology.
Understanding how a protein is put together provides valuable clues to its functions. The development of protein crystallography in the 1930s and 1950s brought several protein structures into view for the first time, but it gradually became apparent that a large fraction of proteins lack a single set structure making them intractable to xray crystallography. As they are too thin for electron microscopy, the only viable alternatives for many of these intrinsically disorderd proteins (IDPs) are nuclear magnetic resonance imaging and small angle xray scattering.
Data collected from these techniques are averaged over ensembles and so give no clear indication of individual protein conformations or how often they occur. Atomic force microscopy on the other hand is capable of nanoscale resolution biological imaging at high-speed, so it can capture dynamics as well as protein structures.
In this latest work researchers at Kanazawa University alongside collaborators in Japan, France and Italy applied the technique to the study of several IDPs and identified parameters defining the shape, size and chain length of protein regions, as well as a power law relating the protein size to the protein length, and a quantitative description of the effect of the mica surface on protein dimensions.
The dynamics of the protein conformations captured thanks to the high-speed capabilities of the technique revealed globules that appear and disappear, and transformations between fully unstructured and loosely folded conformations in segments up to 160 amino acids long.
Studies of the measles virus nucleoprotein in particular helped identify not just the shape and dimensions but also characteristics of the order-disorder transitions in the region responsible for molecular recognition, which allows viruses to identify host factors so that they can reproduce.
They could also determine larger scale structures of the virus’s phosphoprotein that are not accessible to nuclear magnetic resonance (which can only give an indication of distances between amino acides separated by less than 2 nm). The researchers suggest that the formation of certain compact shapes observed may explain the resistance to proteolysis – protein breakdown.
In their report of the work, the researchers highlight that as well as a powerful tool in its own right, “When all molecular features revealed by HS-AFM are combined with the folded local structure given by NMR, the combined information allows a quantitative delineation of the structural and dynamic characters of IDPs, in a more realistic manner compared to the pictures depicted individually, as demonstrated for PNT [measles virus phosphoprotein].”
Background
High-speed atomic force microscopy
Atomic force microscopy was developed in the 1980s and brought the atomic scale resolution achieved by scanning tunnelling microscopy (which won the 1986 Nobel Prize for Physics) to non-conducting samples. It works using a tiny cantilever with a nanoscale tip at the end, which either feels the surface much like a vinyl record needle or taps it. Whether by adjusting the tip height or the resonant frequency of the tapping, the interactions between tip and surface provide a signal that can be used to generate an image.
While AFM images brought huge benefits to biological research, these studies were able to move up a gear again when Toshio Ando and his team at Kanazawa University reported an atomic force microscope that operated at high speed. Atomic scale resolution images became movies bringing not just structures but also dynamics within grasp. Previous work on ordered proteins, which are reasonably well understood, as well as the IDP facilitates chromatin transcription (FACT) protein, has established that the technique can be used to image these biomolecules without effects from contact between tip and sample distorting the data.
Intrinsically disordered proteins
The arrival of xray crystallography gave researchers a clear view of vast numbers of biomolecule structures for the first time. But with the hundreds of thousands of biomolecule structures analysed using protein crystallography since the technique first came into use in the 1930s and 1950s, a mounting body of evidence began to build that not all proteins have a single set structure. The observations ran counter to the prevailing paradigm of protein function determined by a fixed structure.
Over the past ten to twenty years the ubiquity of these intrinsically disorderd proteins and their importance in cell functions from signalling to the regulation of transcription and subsequent translation has become widely recognized. In the current work the researchers study IDPs including polyglutamine tract binding protein-1 (PQBP-1, involved in different processes, such as pre-mRNA splicing, transcription regulation, innate immunity and neuron development), autophagy proteins (which are invovolved in removing dysfunctional cell components) containing intrinsically disordered regions (IDRs) and the measles virus nucleoprotein.
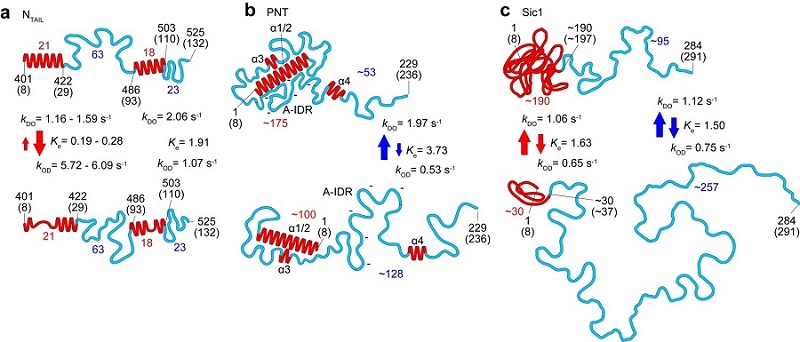
Figure 1. Structural and dynamic features of three IDPs (NTAIL, PNT and Sic1) revealed by HS-AFM imaging. The top and bottom panels correspond to the more-ordered and less ordered states, respectively. The numbers in red and blue represent the numbers of amino acids contained in the respective folded regions and fully disordered regions, respectively. The red and blue arrows indicate the kinetic nature of changes in height of folded regions and end-to-end distance of fully disordered regions, respectively.
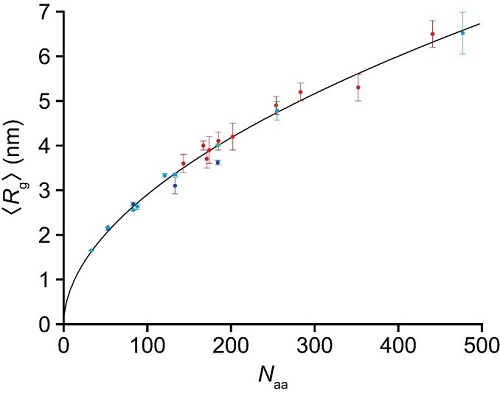
Figure 2. Power law for relationship between radius gyration <Rg> and number of amino acids for 23 fully disordered regions: <Rg> = βg × Naaν (βg = 0.26 nm, and ν = 0.52). The data plotted with the blue circles are those from the present SAXS study for four fully disordered segments of PQBP-1 (82−265, 82−214, 82−164, and 82−134). The nine data plotted with the cyan circles are from the present HS-AFM studies for different IDP constructs. The end-to-end distance of fully disordered regions, <R2D>, observed by HS-AFM was converted to <Rg> using <Rg> = <R2D>/(2√3u), where u = 1.24 was determined by best fitting of all 23 data to a power law, in which the parameter u was contained in the nine data as one of the variables to be determined. The 10 data plotted with the red circles are those from the SAXS study by Mylonas et al. for tau protein constructs [Biochemistry 47, 10345–10353 (2008)], with phosphor-mimic constructs and those significantly affected by the extended repeat domain having been removed here.
Read the original article on Kanazawa University.